Introduction
Protein kinases transfer phosphate groups from ATP to serine, threonine, or tyrosine residues on protein peptide substrates, directly affecting the activity and function of the target. Radiolabel studies suggest that approximately 30% of proteins in eukaryotic cells are subject to phosphorylation.1, 2 This crucial post-translational modification regulates a broad range of cellular activities including the cell cycle, differentiation, metabolism, and neuronal communication. In addition, abnormal phosphorylation events are implicated in many disease states. When assessing phosphorylation, the method of choice may vary depending on many factors including the specific question being asked and availability of specialized equipment or reagents. This review provides a brief description of several methodologies currently used and addresses some of the benefits and drawbacks associated with each.
Kinase Activity Assays
Protein kinases are often common elements in multiple signaling networks influencing numerous downstream effectors responsible for a biological response. Thus, assessing the activity of a single specific kinase may provide valuable information on parallel pathways.3 Kinase activity within a biological sample is commonly measured in vitro by incubating the immunoprecipitated kinase with an exogenous substrate in the presence of ATP. Measurement of the phosphorylated substrate by a specific kinase can be assessed by several reporter systems including colorimetric, radioactive, or fluorometric detection.4 Additionally, R&D Systems offers a non-radioactive Universal Kinase Activity Kit that allows for the quantification of kinase activity for any kinase that produces ADP. Although information can be obtained regarding the actions of a specific kinase, assessing enzyme activity in cellular extracts only provides a glimpse of the signaling landscape. Little is revealed about the proteins being modified, and in vitro activity assays do not address the role of potential endogenous phosphatase activity. Direct detection of phosphorylated proteins can provide a more detailed analysis of the cellular response to an external stimulus, as identification of a phosphopeptide provides information regarding the expression and the functional state of that protein.
Phospho-Specific Antibody Development
A classical method of directly measuring protein phosphorylation involves the incubation of whole cells with radiolabeled 32P-orthophosphate, the generation of cellular extracts, separation of proteins by SDS-PAGE, and exposure to film.2, 5 This labor-intensive method requires many multi-hour incubations and the use of radioisotopes. Other traditional methods include 2-dimensional gel electrophoresis, a technique that assumes phosphorylation will alter the mobility and isoelectric point of the protein.
In light of these laborious methods, the development of phosphorylation-dependent antibodies was a welcome event for researchers. In 1981, the first documented phospho-antibody was produced in rabbits immunized with benzonyl phosphonate conjugated to keyhole limpet hemocyanin (KLH). This antibody broadly recognized proteins containing phosphotyrosine.6 Ten years later, phosphorlyation state-specific (phospho-specific) antibodies were developed by immunizing rabbits with synthetic phosphopeptides representing the amino acid sequence surrounding the phosphorylation site of the target protein.7 The immune sera was applied to a peptide affinity column to generate a highly specific immunoreagent. The availability of phospho-specific antibodies has opened the door for the improvement of traditional methods as well as the development of new immunoassay techniques (View Phospho-specific antibodies). The main caveat in utilizing phospho-specific antibodies in any technique is that successful detection is dependent on the specificity and affinity of the antibody for the phospho-protein of interest.
Western Blot
The Western blot is the most common method used for assessing the phosphorylation state of a protein, and most cell biology laboratories possess the equipment necessary to perform these experiments. Following separation of the biological sample with SDS- PAGE and subsequent transfer to a membrane (usually PVDF or nitrocellulose), a phospho-specific antibody can be used to identify the protein of interest (View Phospho-specific antibodies for Western blot analysis). The typical Western blot protocol eliminates the hazards and waste disposal requirements associated with the use of radioisotopes. Many phospho-specific antibodies are quite sensitive and can readily detect the phosphorylated protein in a routine sample (e.g., 10-30 µg whole cell extract). Because the measured levels of a phospho-protein may change with treatment or through gel loading errors, researchers often utilize an antibody that detects the total level of the cognate protein (regardless of phosphorylation state) to determine the phosphorylated fraction relative to the total fraction and to serve as an internal loading control (Figure 1). Both chemiluminescent and colorimetric detection methods are common, and molecular weight markers are also generally used to provide information about protein mass. A detailed Western blot protocol can be found at www.RnDSystems.com/go/WesternBlotProtocol.
 View Larger Image |
Figure 1. Phosphorylated p53 in CEM Cells. Human T lymphoblast CEM cells were exposed to UV-C light. Cellular extracts generated at 30 or 60 minutes post-irradiation were assessed by Western blot using a Rabbit Anti-Human Phospho-p53 (S15) Polyclonal Antibody (Catalog # AF1043, upper panel) or a Goat Anti-Human p53 Polyclonal Antibody (Catalog # AF1355, lower panel). Indicated samples were treated with lambda-phosphatase (lambda-PPase). |
Simple Western Assays: Automated Capillary Westerns
Simple Western™ instruments provide fully automated capillary-based Western blot analysis and provide several advantages over traditional Western blotting methods for the detection and analysis of protein phosphorylation. Simple Western requires only 3 µL of sample and offers highly sensitive multiplex detection to quantify low abundance phosphorylated and total-protein isoforms in a sample. The Stellar™ NIR/IR Modules for the Simple Western instrument Jess™ provide industry-leading fluorescence detection sensitivity for Western blotting workflows that is 100X more sensitive than traditional Western blot. Jess users can multiplex across NIR/IR fluorescence and chemiluminescence channels with simultaneous Total Protein Normalization for accurate protein expression measurements (Figure 2). Simple Western replaces the strip and re-probe method of traditional Western blots with automated RePlex™ assays, providing sequential immunoassays or total protein detection. In addition to Size-based Simple Western assays, Simple Western Charge assays use capillary isoelectric focusing (cIEF) to resolve all phosphorylated protein isoforms in a sample for detailed and quantitative analysis of protein phosphorylation. Simple Western can process up to 96 samples in a single automated run, enabling the analysis of entire signaling pathways in a single experiment. Learn more about our Simple Western instruments from ProteinSimple, a Bio-Techne brand.
 View Larger Image |
Figure 2. Simple Western multiplex analysis of total AKT and pAKT with simultaneous total protein detection. Lysates from Jurkat cells either untreated (-) or treated (+) with Calyculin A were probed for total AKT (IR, green bands) and phospho-AKT (NIR, red bands). Each antibody was run alone and multiplexed (left panel) in both lysates along with a Stellar Total Protein Assay (right panel). |
Enzyme-Linked Immunosorbent Assay (ELISA)
The ELISA has become a powerful method for measuring protein phosphorylation. ELISAs are more quantitative than Western blotting and show great utility in studies that modulate kinase activity and function. The format for this microplate-based assay typically utilizes a capture antibody specific for the desired protein, independent of the phosphorylation state. The target protein, either purified or as a component in a complex heterogeneous sample such as a cell lysate, is then bound to the antibody-coated plate. A detection antibody specific for the phosphorylation site to be analyzed is then added. These assays are typically designed using colorimetric or fluorometric detection. The intensity of the resulting signal is directly proportional to the concentration of phosphorylated protein present in the original sample (Figure 3). The phospho-specific ELISA technique confers several advantages over more traditional immunoblotting in the measurement of protein phosphorylation. First, results are easily quantifiable by utilizing a calibrated standard. Second, high specificity is possible due to the use of two antibodies specific for the target protein employed together in the sandwich format. Third, the higher sensitivity often accomplished using ELISAs allows for smaller sample volumes and the detection of low abundance proteins. Finally, the microplate-based format allows for much higher throughput than traditional Western blotting (View DuoSet IC ELISA Development Systems). ELISAs generally provide an indirect measurement of kinase activity.
 View Larger Image |
Figure 3. The Human Phospho-EGF R DuoSet IC ELISA is More Sensitive than Immunoprecipitation (IP)-Western Blot Analysis. The A431 human epidermoid carcinoma cell line was treated with 25 ng/mL Recombinant Human EGF (Catalog # 236-EG) for 5 minutes to induce tyrosine phosphorylation of EGF R. Lysates were serially diluted and analyzed by (A) IP-Western blot and (B) the Human Phospho-EGF R DuoSet IC ELISA (Catalog # DYC1095B-2). Immunoprecipitations were performed using an anti-EGF R monoclonal antibody and anti-mouse IgG agarose. Immunoblots were incubated with an HRP-conjugated Anti-Phospho-Tyrosine Monoclonal Antibody (Catalog # HAM1676) to detect phospho-EGF R. Bands were visualized by chemiluminescent detection. Human Phospho-EGF R can be detected with the DuoSet IC ELISA by using approximately 2-4 times less lysate than is needed for a conventional IP-Western blot. |
 View Larger Image |
Figure 4. Use of the Human Phospho-EGF R DuoSet IC ELISA to Quantify Phospho-EGF R in Cells Pretreated with the PD168393 Inhibitor. The A431 human epidermoid carcinoma cell line was left untreated or treated with 25 ng/mL Recombinant Human EGF (Catalog # 236-EG) for 5 minutes, after either being treated without or with 1 nM, 10nM, or 100 nM PD168393. ELISA and IP-Western blot (inset) analyses were performed using 25 and 50 µg of lysate, respectively. IP-Western blots for Phospho-EGF R (p-EGF R) were performed using an anti-EGF R monoclonal antibody and anti-mouse IgG agarose. Immunoblots were incubated with an HRP-conjugated Mouse Anti-Phospho-tyrosine Monoclonal Antibody (Catalog # HAM1676) to detect phosphorylated EGF R. Blots were stripped and total EGF R was detected using a Biotinylated Anti-EGF R Polyclonal Antibody (Catalog # BAF231). ELISA analysis of Phospho-EGF R was performed using the Human Phospho-EGF R DuoSet IC ELISA (Catalog # DYC1095B-2). The DuoSet IC ELISA results correlate well with the relative amount of phosphorylated EGF R detected by IP-Western blot. |
Intracellular Flow Cytometry and ICC/IHC
The traditional techniques of intracellular flow cytometry and immunocytochemistry/immunohistochemistry (ICC/IHC) are powerful tools for detecting phosphorylation events.8, 9 Flow cytometry uses a laser to excite the fluorochrome used for antibody detection. Filter sets and fluorochromes with non-overlapping spectra must be carefully chosen when assessing multiple proteins in the same cell. Flow cytometry is advantageous because it allows for rapid, quantitative, single cell analysis (Figure 5).10 Proteins can be detected in a specific cell type within a heterogeneous population via cell surface marker phenotyping without the need to physically separate the cells. In this way, a small, rare population of cells may be analyzed without concern for cell loss or altered protein expression that may occur during a cell-sorting process (View phospho-specific antibodies for flow cytometry).
 View Larger Image |
Figure 5. Detection of STAT6 in Human Peripheral Blood Mononuclear Cells (PBMCs) by Flow Cytometry. Human peripheral blood mononuclear cells (PBMCs) either (A) Th2-stimulated or (B) unstimulated were stained with a PE-conjugated Rabbit Anti-Human Phospho-STAT6 (Y641) Monoclonal Antibody (Catalog # IC37173P) and an APC-conjugated Mouse Anti-Human CD4 Monoclonal Antibody (Catalog # FAB3791A). Quadrant markers were set based on staining with a PE-conjugated Rabbit IgG Antibody (Catalog # IC1051P). To facilitate intracellular staining, cells were fixed with Flow Cytometry Fixation Buffer (Catalog # FC004) and permeabilized with methanol. |
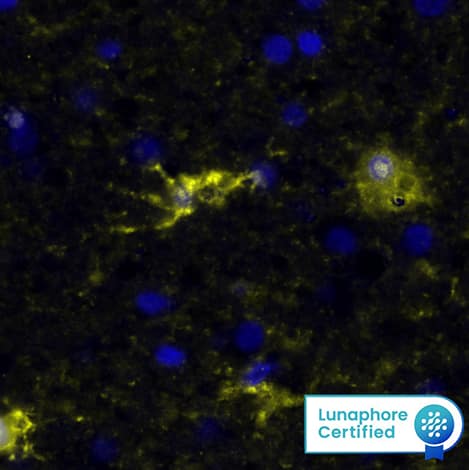
ICC generally refers to protein detection by microscopy in cultured cells, while IHC refers to protein detection in intact tissue sections. Like flow cytometry, these techniques allow for the assessment of multiple proteins within a cell or tissue provided that adequate attention is given to avoid overlapping fluorescence spectra or color. Both fluorescent and colorimetric detection techniques are commonly used (Figure 6). In contrast to other formats for monitoring phosphorylation, ICC is usually the method of choice for determining intracellular localization (View phospho-specific antibodies for ICC/IHC). Both flow cytometry and ICC/IHC require high-affinity and high-specificity antibodies, blocking steps, controls, and antibody titration to eliminate ambiguous results due to non-specific binding.
Detection of phospho-proteins by flow cytometry and ICC require that the protein is stable and accessible to the antibody. Cells are usually stimulated and fixed with formaldehyde or paraformaldehyde to cross-link the phospho-proteins and stabilize them for analysis. The fixed cells must then be permeabilized to allow for entry of phospho-specific antibodies into the cells. Different permeabilization techniques are often useful for various subcellular locations. A mild detergent will allow for detection of cytoplasmic proteins, while alcohol may be required for antibody access to nuclear proteins. Alcohol permeabilization may also enhance phospho-protein detection using peptide specific antibodies due to the denaturing property of alcohol.10 For more detailed ICC/IHC protocols, please visit our website at www.rndsystems.com/protocol-types/iccihc-protocols.
 View Larger Image |
Figure 6. Detection of Phosphorylated Proteins Using ICC/IHC. Phosphorylated ERK1 (T202/Y204) and ERK2 (T185/Y187) were detected in immersion-fixed Hela human cervical epithelial carcinoma cells, (A) unstimulated or (B) stimulated with PMA, using a Rabbit Anti-Human/Mouse/Rat Phospho-ERK1 (T202/Y204)/ERK2 (T185/Y187) Monoclonal Antibody (Catalog # MAB1018) for 3 hours at room temperature. Cells were stained using the NorthernLights™ 557-conjugated Anti-Rabbit IgG Secondary Antibody (red; Catalog # NL004) and counterstained with DAPI (blue; Catalog # 5748). (C) Phosphorylated Tie-2 was detected in immersion-fixed paraffin-embedded sections of human kidney tissue using a Rabbit Anti-Human/Mouse Phospho-Tie-2 (Y992) Antigen Affinity-purified Polyclonal Antibody (Catalog # AF2720) at 1 ug/mL overnight at 4 °C. Before incubation with the primary antibody, tissue was subjected to heat-induced epitope retrieval. Tissue was stained using the Anti-Rabbit IgG VisuCyte™ HRP Polymer Antibody (brown; Catalog # VC003) and counterstained with hematoxylin (blue). Specific staining was localized to the cytoplasm of the convoluted tubules. |
Mass Spectrometry
A comprehensive assessment of protein phosphorylation (phosphoproteomics) in complex biological samples, such as cell lysates, is important for understanding phosphorylation-based signaling networks. Large-scale phospho-protein analysis in complex protein mixtures involves identification of phospho-proteins and phosphopeptides and sequencing of the phosphorylated residues. Mass spectrometry (MS) techniques are useful tools for these tasks. Although MS can be used with excellent sensitivity and resolution to identify a single protein, there are several inherent difficulties for the analysis of phospho-proteins. First, signals from phosphopeptides are generally weaker, as they are negatively charged and poorly ionized by electrospray MS, which is performed in the positive mode.11 Second, it can be difficult to observe the signals from low-abundance phospho-proteins of interest in the high-background of abundant non-phosphorylated proteins. To overcome these drawbacks, several enrichment strategies for phospho-protein analysis by MS have been developed including immobilized metal affinity chromatography (IMAC),12 phosphospecific antibody enrichment,13 chemical-modification-based methods such as beta-elimination of phospho-serine and -threonine,14 and replacement of the phosphate group with biotinylated moieties.15
Multi-Analyte Profiling
Mass spectrometric techniques such as collision-induced dissociation (CID) and electron transfer dissociation (ETD) provide comprehensive parallel analysis of peptide sequences and post-translational modifications such as phosphorylation.16 These techniques are labor-intensive, and strategies for comprehensive phosphorylation analysis may not be needed if particular pathways are of primary interest. This has led to the development of several novel methods for measuring protein phosphorylation of multiple analytes simultaneously. In general, these involve the use of phospho-specific antibodies and membrane-based (View Proteome Profiler Antibody Arrays) detection formats (Box 1, Figures 7 and 8).17,18 The obvious benefit of these assays is that throughput capability is greatly enhanced by bypassing the need for running multiple individual Western blots or traditional ELISA-based assays. These techniques are also known for providing more data while requiring very little sample volume. In trade, protein profiling assays are typically recognized as being less sensitive than their more conventional counterparts due to potential antibody cross-reactivity.
Phospho-Protein Multiplex Assays
Our Phospho-specific Antibody Arrays Dominate Primary Literature
Data from Our Phospho Antibody Arrays Are Published in High Impact Journals*
|
|||||||||||||||||||||||||||||||||||||||||||||


 View Larger Image |
Figure 9. Detection of Human Protein Kinase Phosphorylaton in IGF-I-treated MCF-7 Cells. MCF-7 human breast cancer cells were either left untreated or treated with 100 ng/mL of Recombinant Human IGF-I (Catalog # 291-G1) for 1 hour. The relative level of phosphorylation of 37 different kinases was simultaneously assessed using the Proteome Profiler™ Human Phospho-Kinase Antibody Array Kit (Catalog # ARY003C). |
Conclusion
Assessing protein phosphorylation is often an essential component of the cell biologist's repertoire for understanding intracellular factors underlying cellular activities. Given the important role kinases play, it is critical for researchers to have quality tools for measuring protein phosphorylation and/or kinase activity. Each technique excels in different contexts, and care must be taken to choose the method that best fits the experimental design (Table 1). This review provides a brief glimpse of several of the most widely used methods for assessing protein phosphorylation. Because of a growing demand, methodologies continue to improve, bringing researchers closer to understanding these complex and important processes that ultimately control cellular function.
| Table 1. Methods Used to Detect Protein Phosphorylation using Phospho-Specific Antibodies: A Relative Comparison of Products Offered by R&D Systems. |
||||||||
| Application | Quantitative | Semi- Quantitative |
High Sensitivity | Single Cell Analysis | Protein Localization | Multi- Analyte Profiling |
Dual detection of total and phospho-protein | Labor Intensive |
| Simple Western | X | X | X | X | - | |||
| Western Blot | X | X | + | |||||
| ELISA | X | X | +/- | |||||
| Intracellular Flow Cytometry | X | X | X | X | +/- | |||
| Immunocytochemistry | X | X | + | |||||
| Mass Spectrometry | X | X | X | X | + | |||
| Proteome Profiler | X | X | - | |||||
References
- Sefton, B.M. & S. Shenolikar (1996) Analysis of Protein Phosphorylation. In Ausubel, F.M. et al. (eds): Current Protocols in Molecular Biology, New York: John Wiley & Sons, Inc. 18.1.1-18.1.5.
- Ubersax, J.A. & J.E. Ferrell (2007) Nat. Rev. Mol. Cell Biol. 8:530.
- Ni, Q. et al. (2006) Methods 40:280.
- Johnson, S.A. & T. Hunter (2005) Nat. Methods 2:17.
- de Graauw, M. et al. (2006) Electrophoresis 27:2676.
- Ross, A.H. et al. (1981) Nature 294:654.
- Czernik, A.J. et al. (1991) Methods Enzymol. 201:264.
- Zell, T. et al. (2001) Proc. Natl. Acad. Sci. USA 98:10805.
- Willinger, T. et al. (2005) J. Immunol. 175:5895.
- Krutzik, P.O. et al. (2004) Clin. Immunol. 110:206.
- Mann, M. et al. (2002) Trends Biotechnol. 20:261.
- Brill, L.M. et al. (2004) Anal. Chem. 76:2763.
- Steen, H. et al. (2002) J. Biol. Chem. 277:1031.
- Zhou, H. et al. (2001) Nat. Biotechnol. 19:375.
- Oda, Y. et al. (2001) Nat. Biotechnol. 19:379.
- Molina, H. et al. (2007) Proc. Natl. Acad. Sci. USA 104:2199.
- Stiehl, D.P. et al. (2012) Oncogene 31:2283.
- Bai, Y. et al. (2012) Cancer Res. 72:2501.
 Pan Specific antibody by Western Blot.")











 antibody by Western Blot.")


 antibody by Western Blot.")

 antibody in Human Stomach Cancer Tissue by Immunohistochemistry (IHC-P).")


 antibody by Western Blot.")









 is a blocking buffer for IHC, ICC, ELISA and Western blotting; used to block non-specific antibody binding. Commonly used in cell culture")



 antibody by Western Blot.")